Erweitertes
Neugeborenen-Screening
Erweitertes Neugeborenen-Screening
Das erweiterte Neugeborenenscreening zählt zu den essentiellen sowie erfolgreichen Maßnahmen der Sekundärprävention im Kindesalter. Es dient der frühzeitigen Detektion von seltenen angeborenen Erkrankungen, die unbehandelt zu schwerwiegenden gesundheitlichen Schäden bis hin zum Tod führen können. Die Analyse im Trockenblut findet mittels analytischer (Tandemmassenspektrometrie, Immunoassay) sowie molekulargenetischer (qPCR) Verfahren statt. Zu den Zielerkrankungen des erweiterten Neugeborenenscreenings gehören eine Reihe von Stoffwechselstörungen, Hormonstörungen sowie einige weitere seltene Erkrankungen (s.u.).
Häufigkeit: ca. 1:80.000
Klinische Symptome: Hautveränderungen, Stoffwechselkrisen, zerebrale Krampfanfälle, neurologische Symptome, geistige Behinderung
Ursache: Defekt der Biotinidase (schwerer Defekt: Restaktivität <10%, partieller Defekt: Restaktivität 10-30%)
Nachweis im Screening: Bestimmung der Biotinidase-Aktivität
Methode: Photometrie
Weitere Diagnostik: Biotinidase-Aktivität im Serum, Molekulargenetik
Falsch positive Ergebnisse: Zerstörung der Enzyms auf der Testkarte (z.B. durch Wärme)
Falsch negative Ergebnisse: Z.n. Transfusion
Therapie: Biotinsubstitution
Im Neugeborenen-Screening wird in unserem Labor die klassische Form, der Galaktose-1-Phosphat-Uridyltransferase-Mangel (GalT) sowie der Galaktokinase-Mangel durch Bestimmung der Galaktose und des Galaktose-1-Phosphates erfaßt. Die letztere Bestimmung ist nur möglich, wenn das Neugeborene Galaktose, z.B. in Form von Muttermilch, zu sich genommen hat.
Häufigkeit: 1:40.000
Klinische Symptome: Trinkschwäche, Ikterus, Leberfunktionsstörung
Ursache: Defekt der Galaktose-1-Phosphat-Uridyl-Transferase
Nachweis im Screening: Enzymaktivitätsmessung; Galaktose + Gal-1-Phosphat-Konzentration
Methode: Fluorimetrischer Test (Enzymaktivität), photometrischer Test (Galaktose-Konzentration)
Weitere Diagnostik: Enzymaktivität im Flüssigblut, Mutationsanalyse, Gal-1-Phosphat im Vollblut
Falsch positive Ergebnisse: Zerstörung der Enzyms auf der Testkarte (z.B. durch Wärme oder verlängerte Transportzeit)
Falsch negative Ergebnisse: Z.n. Transfusion
Therapie: lebenslange laktosefreie, galaktosearme Diät
Häufigkeit: 1:10.000
Klinische Symptome: geistige und statomotorische Entwicklungsstörung
Ursache: Defekt der Phenylalaninhydroxylase (selten Tetrahydrobiopterin-Mangel)
Nachweis im Screening: erhöhte Phenylalanin-Konzentration bzw. Erhöhung des Phenylalanin/Tyrosin-Quotienten
Methode: Tandemmassenspektrometrie (MS/MS)
Weitere Diagnostik: Enzmyaktivität, BH4-Test, Mutationsanalyse
Falsch positive Ergebnisse: Leberfunktionsstörungen, Aminosäuren-Infusion
Falsch negative Ergebnisse: bei zeitgerechter Blutentnahme: keine
Therapie: spezifische (Phenylalanin-arme) Diät, ggf. Tetrahydrobiopterin-Substitution
Hyperphenylalaninämie (HPA): Phenylalanin < 600µmol/L, nicht diätpflichtig
Häufigkeit: 1:200.000
Klinische Symptome: Erbrechen, Lethargie, Krampfanfälle, metabolische Enzephalopathie
Ursache: Defekt im Enzymkomplex der verzweigtkettigigen 2-Ketosäure-Dehydrogenase (BCKA-DH)
Nachweis im Screening: Erhöhte Konzentrationen von Leucin/Isoleucin und Valin, Erhöhung des Quotienten Leu+Ile/Phe
Methode: Tandemmassenspektrometrie (MS/MS)
Second-Tier-Test: Allo-Isoleucin-Bestimmung mittels LC-MS/MS
Weitere Diagnostik: quantitative Aminosäurenanalyse, organische Säuren im Urin, Enzymaktivität in Fibroblasten, Mutationsanalyse
Falsch positive Ergebnisse: Aminosäuren-Infusion
Falsch negative Ergebnisse: milde Formen
Therapie: immer über pädiatrische Stoffwechselzentren
Häufigkeit: 1:135.000 (Zentraleuropa)
Klinische Symptome: schweres Leberversagen, Blutungen, Erbrechen, Sepsis, Nierenfunktionsstörungen
Ursache: Defekt im Abbau der Aminosäure Tyrosin (defektes Enzym: Fumarylacetacetat-Hydrolase (FAH))
Nachweis im Screening: Erhöhte Konzentration von Succinylaceton, das bedingt durch den Rückstau von Maleylacetacatat und Fumarylacetacetat vermehrt gebildet wird
Methode: Tandemmassenspektrometrie (MS/MS)
Weitere Diagnostik: Succinylaceton im Urin, Trockenblut, Serum, Fruchtwasser, quantitative Aminosäurenanalyse, organische Säuren im Urin, Gerinnungsparameter, Transaminasen, AFP, molekulargenetische Untersuchung; im weiteren Verlauf: Succinylaceton und das Medikament NTBC (Orfadin®), Aminosäuren (Met, Tyr, Phe)
Falsch positive Ergebnisse: Nierenfunktions-Störungen
Falsch negative Ergebnisse: milde Formen, akzelerierte Form der Tyrosinämie Typ I
Therapie: immer über pädiatrische Stoffwechselzentren
Häufigkeit: 1:10.000
Klinische Symptome: Stoffwechselkrisen, Koma, letaler Verlauf möglich
Ursache: Defekt der Medium Chain Acyl-CoA-Dehydrogenase
Nachweis im Screening: typisches Metabolitenmuster mit Konzentrationserhöhung der mittelkettigen Acylcarnitine sowie Erhöhung der Quotienten C8/C2, C8/C10, C8/C12
Methode: Tandem-Massenspektrometrie (MS/MS)
Weitere Diagnostik: erneut TMS-Profil, Enzymaktivität in Lymphozyten, Molekulargenetik.
Falsch positive Ergebnisse: ggf. bei MCT-Gabe, Valproat-Therapie
Falsch negative Ergebnisse: bei ausgeglichener und anaboler Stoffwechsellage möglich
Therapie: Vermeidung von Fastenperioden >4-6 Std., ggf. Carnitingabe
Häufigkeit: ca 1:200.000
Klinische Symptome: Manifestation schon in den ersten Lebenstagen möglich, Stoffwechselkrisen, Koma, Kardiomyopathie, Hepatopathie. In einigen Fällen mütterliches HELLP-Syndrom.
Ursache: Defekt der Long-Chain-3-OH-Acyl-CoA-Dehydrogenase (LCHAD) bzw. des trifunktionellen Proteins (TFP)
Nachweis im Screening: typisches Metabolitenmuster mit Konzentrationserhöhung der langkettigen hydroxylierten Acylcarnitine (v.a. C16-OH, C18:1-OH)
Methode: Tandem-Massenspektrometrie (MS/MS)
Weitere Diagnostik: Enzymaktivität in Fibroblasten oder Lymphozyten, Molekulargenetik.
Falsch negative Ergebnisse: bei anaboler Stoffwechsellage möglich
Therapie: Mittelkettige Triglyceride, Vermeidung von Fastenperioden, reduzierte Aufnahme langkettiger Fettsäuren
Häufigkeit: ca. 1:100.000
Klinische Symptome: Kardiomyopathie, Hepatopathie, hypoketotische Hypoglykämie, Skelettmuskelbeteiligung. Milde Form: Spätmanifestation mit Myopathie
Ursache: Defekt der (über-)langkettigen Acyl-CoA-Dehydrogenase (veryl long chain acyl CoA dehydrogenase deficiency, (VLCADD))
Nachweis im Screening: Konzentrationserhöhung C14:1, Quotient C14:1/C12:1 erhöht
Methode: Tandemmassenspektrometrie (MS/MS)
Weitere Diagnostik: Enzymaktivität in Fibroblasten oder Lymphozyten, Molekulargenetik.
Anmerkung: ein unauffälliger Befund im Neugeborenen-Screening schließt das Vorliegen eines VLCAD-Mangels nicht aus!
Therapie: Mittelkettige Triglyceride, Vermeidung von Fastenperioden, reduzierte Aufnahme langkettiger Fettsäuren
Häufigkeit: selten
Klinische Symptome: hypoketotische Hypoglykämie, Krampfanfälle, Koma, Hepatopathie, renal-tubläre Azidose. Manifestation i.A. innerhalb der ersten zwei Lebensjahre
Ursache: Defekt der Carnitin-Palmitoyl-Transferase 1 (gestörter Transport der langkettigen Acylcarnitine in die Mitochondrien)
Nachweis im Screening: freies Carnitin erhöht, Konzentration der langkettigen Acylcarnitine vermindert: Quotient Carnitin/[C16+C18] erhöht.
Methode: Tandemmassenspektrometrie (MS/MS)
Weitere Diagnostik: Enzymaktivität in Fibroblasten, Molekulargenetik. Organische Säuren im Urin: keine Dicarbonazidurie
Therapie:Mittelkettige Triglyceride, Vermeidung von Fastenperioden, reduzierte Aufnahme langkettiger Fettsäuren, Notfallregime
Häufigkeit: selten
Klinische Symptome: Hypoketotische Hypoglykämie, Hepatopathie, hepatische Enzephalopathie, Krampfanfälle. Verschiedene Verlaufsformen (schwere neonatale, infantile sowie milde Erwachsenenform
Ursache: Defekt der Carnitin-Palmitoyl-Transferase 2
Nachweis im Screening: erhöhte Konzentration der langkettigen Acylcarnitine. Quotient [C16+C18:1]/C2 erhöht
Methode: Tandemmassenspektrometrie (MS/MS)
Differenzialdiagnose: CACT-Mangel
Weitere Diagnostik: Enzymaktivität in Lymphzyten oder Fibroblasten. Mutationsanalyse. Organische Säuren: keine bzw unspezifische Dicarbonazidurie.
Therapie: Mittelkettige Triglyceride, Vermeidung von Fastenperioden, reduzierte Aufnahme langkettiger Fettsäuren, Notfallregime. Intensivtherapie bei schwerer neonataler Form
Häufigkeit: selten
Klinische Symptome: Lethargie, Trinkschwäche, Kardiomyopathie, Hepatopathie, Hyperammonämie. Häufig lethaler Verlauf
Ursache: Defekt der Carnitin-Acylcarnitin-Translokase
Nachweis im Screening: erhöhte Konzentration der langkettigen Acylcarnitine, Quotient [C16+C18:1]/C2 erhöht
Methode: Tandemmassenspektrometrie (MS/MS)
Differenzialdiagnose: CPT2-Mangel
Weitere Diagnostik: Enzymaktivität in Lymphozyten oder Fibroblasten. Mutationsanalyse.
Therapie: Intensivtherapie bei schwerer Form. Mittelkettige Triglyceride, Vermeidung von Fastenperioden, reduzierte Aufnahme langkettiger Fettsäuren
Häufigkeit: ca. 1:100.000
Klinische Symptome: Makrozephalie, enzephalopathische Krise; in den ersten Lebensmonaten oft unauffällig.
Ursache: Defekt der Glutaryl-CoA-Dehydrogenase
Nachweis im Screening: erhöhte Glutarylcarnitinkonzentration (C5DC)
Methode: Tandemmassenspektrometrie (MS/MS)
Weitere Diagnostik: Carnitin und Acylcarnitine im Plasma, organische Säuren im Urin, Enzymaktivität in Lymphozyten oder Fibroblasten, Mutationsanalyse
Therapie: Carnitingabe, Notfallprotokoll, spezifische Diät
Falsch negative Ergebnisse: bei Carnitinmangel sowie abhängig von der Stoffwechsellage möglich.
Häufigkeit: ca 1:100.000
Klinische Symptome: neonatale Form: akute Dekompensation mit enzephalopathischer Erkrankung. Chronisch-intermittierende Form mit Episoden von Erbrechen, Lethargie, Koma.
Ursache: Defekt der Isovaleryl-CoA-Dehydrogenase
Nachweis im Screening: erhöhte Isovalerylcarnitin-Konzentration (C5)
Methode: Tandemmassenspektrometrie (MS/MS)
Weitere Diagnostik: Carnitin und Acylcarnitine im Plasma, organische Säuren im Urin, Enzymaktivität in Lymphozyten oder Fibroblasten, Mutationsanalyse
Therapie: Carnitingabe, Notfallprotokoll, spezifische Diät
Differenzialdiagnose: 2-Methylbutyryl-CoA-Dehydrogenase-Mangel
Falsch positive Ergebnisse: C5-Erhöhung durch Pivaloylcarnitin bei Gabe Pivalinsäure-haltiger Antibiotika (in Deutschland nicht eingesetzt, da nicht zugelassen)
Häufigkeit: ca 1:4000
Klinische Symptome: (konnatale Hypothyreose): schwere geistige und körperliche Behinderung.
Ursache: Fehlanlage der Schiddrüse (Aplasie, Hypoplasie, Ektopie), selten genetisch bedingt.
Nachweis im Screening: TSH (Thyreoida stimulierendes Hormon) stark erhöht.
Methode: Immunoassay
Weitere Diagnostik: TSH, fT3, fT4 im Serum. Schilddrüsensonographie. (Vergl.Angeborene primäre Hypothyreose: Diagnostik, Therapie und Verlaufskontrolle“ auf den Webseiten der AWMF)
Falsch positive Ergebnisse: Lebensalter <36 Std: Der Geburtsstress (Kältereiz) führt zu einem vorübergehenden TSH-Anstieg. Transiente Hypothyreose durch Jodbelastung (jodhaltige Desinfektionsmittel, jodhaltige Röntgenkontrastmittel), Therapie mit Thyreostatika in der Schwangerschaft, Übertragung mütterlicher Schilddrüsenantikörper über die Plazenta.
Falsch negative Ergebnisse: unter Katecholamingabe. Hypothyroxinämie bei Frühgeborenen (<32 SSW).
Therapie: Thyroxin-Substitution
Häufigkeit: ca. 1:10.000
Klinische Symptome: verminderte Stresstoleranz, Virilisierung bei Mädchen, Salzverlustkrise (Erbrechen, Exsikkose, Hyperkaliämie, Azidose, Hyponatriämie)
Ursache: 21-Hydroxylase-Mangel (ca.95% der Fälle).
Nachweis im Screening: 17-Hydroxyprogesteron deutlich erhöht.
Methode: Immunoassay
Weitere Diagnostik: Second-Tier-Test: Steroidprofil mittels LC-MSMS. Molekulargenetik
Falsch positive Ergebnisse: Lebensalter <36 Std., Stress (schwere Erkrankung), Frühgeburtlickeit
Falsch negative Ergebnisse: Steroidhormongabe während der Schwangerschaft
Therapie: Hormonsubstitution
Häufigkeit: ca. 1:58.000
Klinische Symptome: lebensbedrohliche, schwer verlaufende Infektionen in den ersten Lebenstagen kurz nach Geburt
Hinweis: Bei fehlenden TREC (Kopienzahl 0 oder 1, urgent positiv) handelt es sich um einen immunologischen Notfall!
Ursache: schwerer Mangel an T-zellen und naiver T-Zellen
Nachweis im Screening: verminderte oder fehlende T-cell receptor excision circles (TREC)
Methode: Polymerase-Kettenreaktion (PCR)
Weitere Diagnostik: qualifizierte FACS-Analyse in einem zertifizierten Zentrum (siehe Arbeitsgemeinschaft Pädiatrische Immunologie e.V.: www.kinderimmunologie.de)
Falsch positive Ergebnisse: Frühgeburtlichkeit, andere Erkrankungen, syndromale Erkrankungen, weitere T-Lymphopenien
Falsch negative Ergebnisse: atypische Formen
Therapie: Isolation, allogene Stammzelltransplantation
Häufigkeit: ca. 1:7.000
Klinische Symptome: Muskuläre Hypotonie (floppy infant), proximal betonte Muskelschwäche, Verlust motorischer Fähigkeiten, auffallendes Atemmuster mit Zwerchfellatmung, Glockenthorax, Muskelzucken (Faszikulationen, Zungenfaszikulationen), reduzierte Ausdauer als erstes Symptom einer Muskelschwäche, auffälliges Gangbild, Muskelkrämpfe Muskelatrophie; Einteilung in unterschiedliche Schweregrade der Erkrankung und des Erkrankungsbeginns (SMA 1-4; non-Sitter, Sitter, Walker)
Hinweis: Eine frühzeitige Bestätigung des Ergebnisses aus Vollblut sowie die zeitnahe Vorstellung des Kindes in einem empfohlenen Behandlungszentrum ist notwendig
Ursache: homozygote Deletion der Exons 7 und 8 oder nur des Exons 7 des Survival Motor Neuron 1 (SMN1) Gens auf Chromosom 5q13.2 (ca. 95% aller Fälle)
Nachweis im Screening: Homozygotie des SMN1-Gens
Methode: Polymerase-Kettenreaktion (PCR)
Weitere Diagnostik: molekulargenetische Diagnostik, Bestimmung der SMN2-Kopienanzahl (kann aus vorhandener Trockenblutprobe nachgefordert werden)
Falsch positive Ergebnisse: Störungen der PCR-Reaktion
Falsch negative Ergebnisse: atypische Formen, eine Compound-Heterozygotie für die Deletion und eine Punktmutation im SMN1-Gen (ca. 4%) kann aus methodischen Gründen im Neugeborenen-Screening nicht erkannt werden
Therapie: je nach Anzahl der Kopien und der Schwere der Erkrankung wird eine individuelle Therapie (medikamentös, Genersatztherapie) festgelegt
Häufigkeit: 1-5:10.000
Klinische Symptome: chronische hämolytische Anämie, erhöhte Infektneigung, rezidivierende schmerzhafte Gefäßverschlusskrisen, chronische Organschädigungen
Hinweis: Bestätigung des auffälligen Screening-Ergebnisses aus Vollblut innerhalb des ersten Monats nach Geburt in einem empfohlenen Behandlungszentrum ist notwendig
Ursache: Punktmutation (Glutamat → Valin an Aminosäureposition 6) im Hämoglobinmolekül führt zur Ausbildung von defekten β-Ketten des Hämoglobins, Bildung von Hämoglobinmolekülen(HbS) statt des physiologischen Haupthämoglobins A (HbA)
Nachweis im Screening: HbS homozygot, compound-heterozygote Formen (FSC, FSD, FSE, HbS + β+-Thalassämie)
Methode: Polymerase-Kettenreaktion (PCR), HPLC, Tandem-Massenspektrometrie
Weitere Diagnostik: Hämoglobinanalyse (Kapillarelektrophorese, HPLC), Molekulargenetik, Sequenzierung
Falsch positive Ergebnisse: Störungen der PCR-Reaktion
Falsch negative Ergebnisse: Erythrozyten-Transfusion, Frühgeburtlichkeit
Therapie: Transfusionen, Infektionsprophylaxe (Penicillin, Impfungen), Stammzelltransplantation
Häufigkeit: ca. 1:5.300 – 1:100.000
Klinische Symptome: psychomotorische Symptome, Retardierung, Gedeihstörungen, atrophische Glossitis, megaloblastäre Amämie, progressive Neuro-. Myo-, Enzelopathie
Ursache: Defekt des Vitamin B12- und Cobalamin-Stoffwechsels (Intrinsic Faktormangel, Transcobalamin I- und II-Mangel, intestinale Cbl-Malabsorption (Imerslund-Gräsbeck-Syndrom)
Nachweis im Screening: Acylcarnitine, Aminosäuren
Methode: Tandem-Massenspektrometrie
Weitere Diagnostik: organische Säuren im Urin, Vitamin B12 und Holotranscobalamin im Plasma
Falsch positive Ergebnisse: mütterlicher (Maternaler) Vitamin B12-Mangel
Falsch negative Ergebnisse: Z.n. Transfusion, FFP, Vitamin B12-Einnahme
Therapie: Holotranscobalamin, Vitamin B12, Folat
Hinweis: Es ist möglich, dass durch das auffällige Ergebnis beim Neugeborenen ein Vitamin B12-Mangel der Mutter entdeckt wird, daher soll bei Kontrolluntersuchungen immer auch das Blut der Mutter mit untersucht werden.
Häufigkeit: ca. 1:170.000 – 1:500.000
Klinische Symptome: progrediente geistige Behinderung, neurologische und psychiatrische Symptome, Thromboembolien, megaloblastäre Anämie, Überstreckung der Gelenke, Bindegewebsschwäche, progrediente Myopie, Epilepsie
Ursache: Cystathionin-beta-Synthase-Mangel
Nachweis im Screening: Aminosäuren Methionin (Met), Homocystein (Hcys)
Methode: Tandem-Massenspektrometrie
Weitere Diagnostik: Aminosäuren im Plasma, Methionin, Homocystein, Cystein
Falsch positive Ergebnisse: Methionin-Synthesestörungen, Cobalaminstörungen
Falsch negative Ergebnisse: Z.n. Transfusion, Frühabnahme, milde Formen
Therapie: Methionin-arme Diät, Pyridoxin, Folsäure, Betain, Hydroxycobalamin, Vitamin C
Häufigkeit: ca. 1:30.000 -1:130.000
Klinische Symptome: geistige Behinderung, Bewegungsstörungen, chronische Nierenfunktionsstörung, Kardiomyopathie,
Ursache: PA: Defekt der Propionyl-CoA-Carboxylase; MMA: Methyl-Malonylmutase-Mangel
Nachweis im Screening: Bestimmung der Acylcarnitine
Methode: Tandem-Massenspektrometrie
Weitere Diagnostik: organische Säuren im Urin, Carnitin-Status, Aminosäuren im Plasma
Falsch positive Ergebnisse: Niereninsuffizienz, bakterielle Endprodukte im Darm
Falsch negative Ergebnisse: Z.n. Transfusion, milde Formen
Therapie: PA: Valin- und Isoleucin-arme Diät, Carnitin, Lebertransplantation, MMA: Vitamin B12, Carnitin,, PA + MMA: Anbindung an Stoffwechselzentren erforderlich
Screening auf Mukoviszidose/Cystische Fibrose (CF)
Seit dem 1.9.2016 ist zusätzlich zum erweiterten Neugeborenen-Screening das Screening auf Mukoviszidose Bestandteil der Vorsorge-Untersuchungen für Neugeborene entsprechend der aktuell gültigen Kinderrichtlinie. Ab dem 1.1.2017 wird es auch für alle Kinder im ambulanten Bereich durchgeführt und von den Krankenkassen bezahlt. Jedes gesetzlich versicherte Kind hat einen Anspruch auf die Durchführung der Untersuchung.
Die Mukoviszidose ist eine angeborene Stoffwechselerkrankung mit einer Inzidenz von etwa 1 : 3.300. Sie wird autosomal-rezessiv vererbt. Betroffen ist das Protein „Cystic Fibrosis Transmembrane Conductance Regulator“ (CFTR), welches in Zellmembranen lokalisiert ist. Es regelt als Kanalprotein den Wasser- und Chlorid-Transport der Zellen.Bei Veränderung des CFTR-Gens kommt es zur Ansammlung zähen Schleims in diversen Organen, vor allem des Pankreas und der Lunge, aber auch andere Organe können betroffen sein. Die Folge sind schwere chronische Störungen der Lunge und des Pankreas, die die Entwicklung der Betroffenen stark einschränken und im weiteren Verlauf die Lebenswerwartung einschränken können. Das frühzeitige Erkennen der Erkrankung ermöglicht es, die Kinder frühzeitig mit einer Therapie zu versorgen und somit den Progress der Erkrankung zu verzögern.
Das Screening auf Mukoviszidose wird in einem bis drei verschiedenen Tests (siehe Abb.) aus der gleichen Trockenblutkarte durchgeführt, die für die Durchführung des erweiterten Neugeborenen-Screenings verwendet wird. Voraussetzung sind die ausreichende Blutmenge und -Qualität der Blutstropfen auf der Trockenblutkarte.
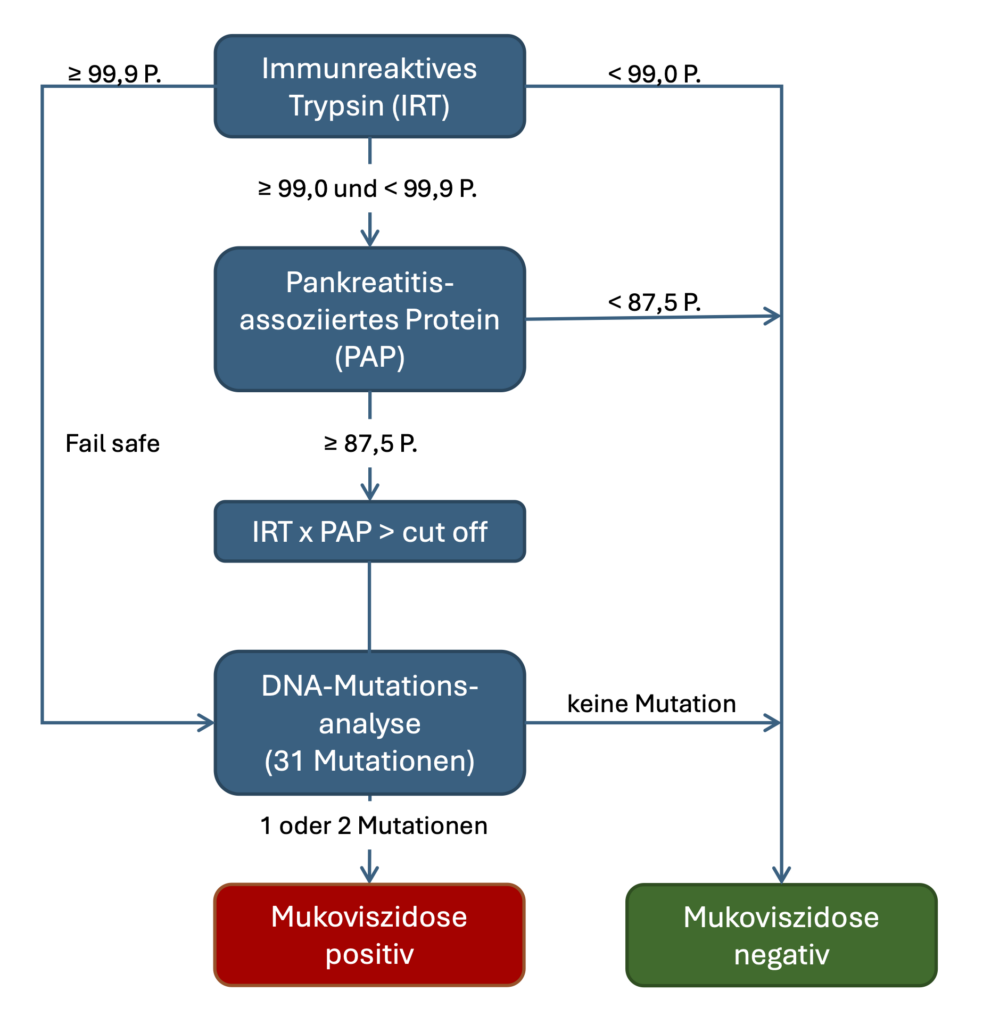
Screening-Ablauf entsprechend der Kinderrichtlinie; Copyright Screening-Labor Hannover 2026
Tracking
Das Tracking, welches ebenfalls in unserem Labor durchgeführt wird, dient der Nachverfolgung auffälliger Befunde. Dazu werden Informationen zu den sich anschließenden Kontrolluntersuchungen und der ggf initiierten Konfirmationsdiagnostik eingeholt, bis entweder eine klare klinische Diagnose festgelegt und die entsprechende Therapie und suffiziente Betreuung eingeleitet wurde, oder ein Krankheitsverdacht ausgeschlossen werden konnte.
Die dabei gewonnenen Daten werden unter ärztlicher Verantwortung und Schweigepflicht für den jährlichen Qualitätsreport an die Deutsche Gesellschaft für Neugeborenenscreening (DGNS) anonymisiert weitergeleitet und dienen gleichzeitig der Qualitätssicherung der postanalytischen Phase.